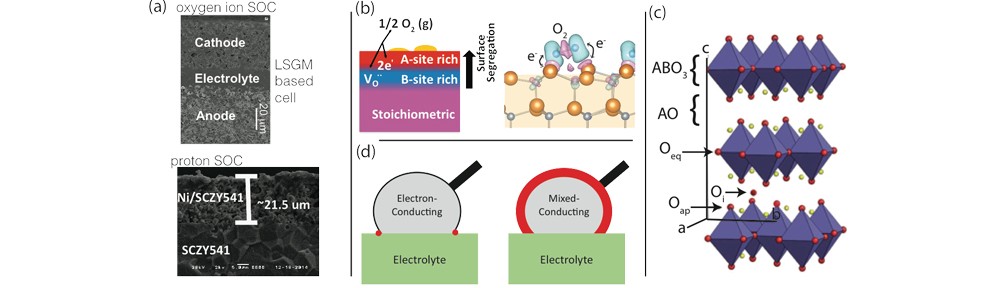
To overcome the challenges posed by the development of high-activity electrodes, we have divided this theme into three scientific opportunities:
B1: Mitigating segregation of cations to the surface of electrodes: What is the impact of surface chemistry on gas exchange kinetics and mechanism?
Many electrode materials are based on the ABO3 perovskite structure, which often have the A-site occupied by lanthanum (La3+) but are doped with strontium (Sr2+) to introduce both electron holes and oxygen vacancies. Numerous recent investigations have linked evidence of strontium segregation to the surface, and to the degradation of the electrode. Therefore, it is important to understand the transport of cation species such as Sr under the influence of substantial oxygen potential and electric potential gradients, as well as the role the surface chemistry plays in electrode function. We will carry out a systematic assessment of factors that impact segregation rates experimentally and computationally.
B2: Designing new Ruddlesden-Popper structured electrodes for enhanced durability and increased activity.
An alternative, promising route to avoiding cation segregation issues described in Opportunity B1 is to find materials that do not require high levels of substitution of elements such as Sr to achieve high cathode activity. This has led to interest in the Ruddlesden-Popper (RP) series of materials such as La4NiO4 and Pr2NiO4, which show high electronic conductivity and have the potential to be good mixed conductors.
B3: Identifying new electrodes for proton transport SOECs with mixed proton and electronic conductivity.
We will use an integrated computational/experimental approach to rapidly accelerate the rate of discovery of materials that simultaneously support the coexistence of proton and hole conductivity in oxides. First principles computational approaches similar to those used in Opportunity A2, in which the tendencies for hydration to occur at the site of oxygen ion vacancies will be established, will be applied here as well. Likewise, we will derive conditions for the coexistence of protons and holes in oxide materials. Defect formation energies, computed ab initio, and thermodynamic considerations will be used to establish equilibrium profiles of proton and hole concentrations in candidate materials.
<Back to Research Page